Clinical Characteristics and Treatment Outcome of Waldenstrom Macroglobulinemia: Experience from a Tertiary Cancer Centre
Download
Abstract
Background and objective: Waldenström macroglobulinemia (WM), also known as lymphoplasmacytic lymphoma, is a rare B-cell lymphoproliferative malignancy characterized by the presence of serum monoclonal immunoglobulin M (IgM) protein and lymphoplasmacytic infiltration in the bone marrow. Patients typically present in their seventh decade with symptoms related to bone marrow infiltration or the effects of monoclonal IgM in the bloodstream. This study aimed to analyze the clinical characteristics and treatment outcomes of patients diagnosed with WM.
Materials and methods: This retrospective analysis was conducted on 26 cases of WM treated at a tertiary cancer center.
Results: WM constituted 0.68% of our non-Hodgkin's lymphoma cases. The median age at presentation was 67 years, with a male-to-female ratio of 2:1. Four patients had lymphadenopathy, six had splenomegaly, and four exhibited hyperviscosity. All patients displayed IgM paraproteinemia; the M band was IgM-kappa in 19 patients and IgM-lambda in seven. All patients received histopathological confirmation. According to the International Prognostic Scoring System for WM (IPSS-WM), 12 patients were classified as intermediate risk, 12 as high risk, and 2 as low risk. Among 26 patients, 20 received upfront treatment, while four were initially observed. Treatment indications included cytopenia in ten patients, constitutional symptoms in five, hyperviscosity in four, and symptomatic lymphadenopathy in one. Sixteen patients received rituximab-based chemotherapy, and four patients with hyperviscosity underwent plasmapheresis. The three-year progression-free survival and overall survival rates were 69.4% and 78%, respectively.
Conclusion: WM is a low-grade B-cell lymphoproliferative disorder with an indolent course, often requiring treatment only after prolonged periods. Diagnosis can be challenging due to the lack of distinct diagnostic features. Rituximab-containing regimens represent the standard of care. Newer targeted treatment options hold promise for improving outcomes in this incurable disease.
Introduction
Waldenstroms macroglobulinemia (WM) or Lymphoplasmacytic lymphoma (LPL) is a rare B cell lymphoproliferative malignancy which accounts for accounts for 1.9% of all non Hodgkin’’s lymphoma (NHL). [1]. It is characterized by monoclonal immunoglobulin M (IgM) serum protein and monoclonal lymphoplasmacytic cells in the bone marrow. It is a disease of the elderly, with a median age at diagnosis of 73 years [1]. It is an indolent disease with a median survival ranging from 5 to 10 years or more [2, 3].
Monoclonal gammopathy is an incidental finding seen in about 3.5% of individuals above 50 years of age and WM is an important differential diagnosis of monoclonal gammopathy [4].
The disease presents with symptoms related to the infiltration of bone marrow by small lymphoid cells and the elevated serum IgM paraprotein which produces hyperviscosity and autoimmune phenomena [5, 6].
WM is a diagnosis of exclusion and made only after excluding other small B cell neoplasms. The most important risk factor for developing WM is a preexisting IgM monoclonal gammopathy of undetermined significance (MGUS) which can transform to WM at a rate of 1.5% per year [7].
In this report we present the results of a retrospective analysis of 26 cases of WM treated at a tertiary cancer centre.
Materials and Methods
This is a retrospective study conducted in the Department of Medical Oncology at a tertiary cancer centre in India during a seven-year period (January 2013 to December 2019). Patients with WM above 14 years of age diagnosed during the study period were included. Twenty-six patients were diagnosed with WM during the study period. The study was approved by the Human ethics Committee (HEC No. 40/22). All procedures were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration.
Inclusion/ Exclusion Criteria
Eligibility criteria included all patients aged above 14 years with a histological diagnosis of WM/LPL. Patients with relapsed WM and who received some prior treatments were excluded from the study.
Methodology
Medical records of patients were studied with respect to the demographic details, clinical history, and physical examination, complete hemogram, serum chemistries, beta 2 microglobulin and serum lactate dehydrogenase (LDH). Diagnostic work up including immunoglobulin assay, serum protein electrophoresis (SPE), immunofixation electrophoresis (IFE), serum viscosity, histopathology, bone marrow study, and imaging studies were noted. International prognostic staging for WM (IPSS-WM) was used for staging [3]. The standard treatment for WM included systemic chemo-immunotherapy with the anti-CD 20 monoclonal antibody rituximab and other chemotherapy drugs in varying combinations for 6 cycles. Plasmapheresis or plasma exchange was considered for patients with evidence of hyper viscosity. The treatment responses were obtained at regular intervals during chemotherapy, by end of chemotherapy, at relapse and while on follow up. Clinical response was classified as complete response (CR), Very Good Partial Response (VgPR), partial response (PR), stable disease (SD), and progressive disease (PD) based on response criteria adopted at the 6th International Workshop on WM [8]. Overall survival (OS) was assessed from the initiation of definitive chemotherapy to the last follow up or death and progression free survival (PFS) was calculated from the initiation of chemotherapy till disease progression.
Statistical analysis
The baseline patient characteristics, treatment and response details were analyzed using descriptive statistics. OS and PFS were assessed by Kaplan – Meier method, using SPSS v. 11. The risk for OS and PFS were analysed using Cox regression method. Statistical significance was defined as a p value < 0.05.
Results
Baseline patient characteristics are described in Table 1.
| Baseline patient characteristics (n=26) | Frequency, n (%) |
| Median age (years) | 67 (range 42-88) |
| Age > 65 years | 18 (69 %) |
| M: F ratio | 2:01 |
| B symptoms | 10 (38) |
| The median duration of symptoms (weeks) | 12 (range 1 – 24) |
| Lymphadenopathy | 4 (15) |
| Splenomegaly | 4 (15) |
| Hyper viscosity (> 4 centipoise) | 4 (15) |
| Hb < 10 g/ dL | 13 (50) |
| Platelet count< 100000/mm3 | 04 (15) |
| Elevated LDH | 13 (50) |
| Elevated Beta 2 macroglobulin | 14 (54) |
| M band > 4 g/dL | 6 (23) |
| IFE: IgM-kappa | 19 (73) |
| IgM-Lambda | 7 (27) |
| IPSS –WP scoring | |
| Low risk | 2 (7.7) |
| Intermediate risk | 12 (46.15) |
| High risk | 12 (46.15) |
n, number of patients; M:F ratio, Male: Female ratio, B symptoms, fever/weight loss/night sweats; Hb, Hemoglobin; LDH, Lactate Dehydrogenase; M band, Monoclonal band in serum protein electrophoresis; IFE, Immunofixation Electrophoresis. IPSS International prognostic staging system for WM
During the period 2013 to 2019, 3819 cases of NHL above 14 years of age were treated in our department, of which 26 were WM (0.68%). The median age at presentation was 67 years (range 42-88 years). There were 18 males and 9 females, with the male to female ratio of 2:1. Only two patients were less than 60 years of age (8%) and elderly patients above>65 years constituted 2/3rd (17 patients) of our study population. The most common presenting symptoms were dyspnea on exertion, fatigue, and low backache. One patient each presented with abdominal discomfort, neck swelling, recurrent urinary tract infection, lower limb pain, jaundice and fever. B symptoms were present in 10 patients (38%). The median duration of symptoms was 12 weeks (range 1 – 24 weeks). Four patients had lymphadenopathy (15%), six had splenomegaly (23%) and four patients (15%) had evidence of hyper viscosity at presentation. The mean hemoglobin in the study population was 9.5 g/dL (range 5.6- 15.3g/dL) and four patients had thrombocytopenia less than 100000/mm3 (range 14000-602000/mm3). The mean serum viscosity was 3.5 centipoises (range 1.7- 7.3). Among our patients, one tested positive for tuberculosis. All patients had IgM paraproteinemia on SPE with a median monoclonal band (M band) of 2.6 gm/dL (range 0.8 - 5.9 gm/dL). On IFE the M band was IgM-kappa (IgM-k) in 19 patients and IgM-lambda (IgM-λ) in seven patients. Serum LDH was elevated in 13 patients (50%) and serum beta 2 microglobulin was >4 mg/L in 14 patients (54%).
All patients had a histopathological confirmation either from bone marrow or from a lymph node biopsy. Histopathological examination of bone marrow specimens showed infiltration by plasmacytoid cells in variable numbers mixed with frequent reactive mast cells. On immunohistochemically (IHC) the tumor cells were positive for CD20, surface IgM, CD 138 and negative for CD5, CD10, CyclinD1. Tumor cells showed either k or λ light chain restriction (Figure 1 a,b,c,d).
Figure 1. a) hematoxylin and eosin staining of the bone marrow specimen showing diffuse interstitial infiltration by atypical lymphoplasmacytic cells. (400X). On immunohistochemistry these atypical cells are positive for b) CD138, c) CD 20, and d) shows kappa light chain restriction.

According to IPSS-WM, 12 patients each were in the intermediate and high risk group and 2 in the low risk group.
Treatment
Among the 26 patients, 20 (77%) patients received upfront treatment and two patients refused treatment. Treatment details are summarized in Table 2.
| Treatment outcome | RCD | BR | BRD | RCHOP/RCVP | Rituximab single agent | Others | Total number |
| Number of patients | 6 | 3 | 3 | 2 | 2 | 4 | 20 |
| Complete remission | 4 | 3 | 2 | 2 | 1 | 1 | 13 |
| Partial remission | 0 | 0 | 0 | 0 | 1 | 1 | 2 |
| Progressive disease | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Death | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Relapse | 0 | 0 | 1 | 0 | 1 | 1 | 3 |
| Defaulted | 1 | 0 | 1 | 0 | 0 | 1 | 3 |
RCD, Rituximab + cyclophosphamide + dexamethasone; BR, Bendamustine + Rituximab; BRD, Bortezomib +rituximab + dexamethasone; R-CHOP/RCVP, Rituximab + Cyclophosphamide + Vincristine + doxorubicin + Prednisolone/ Rituximab + Cyclophosphamide + Vincristine + Prednisolone.
Four patients with WM had no constitutional symptoms, no anemia, no symptoms attributable to IgM monoclonal protein or tumor infiltration at the time of presentation. These patients were diagnosed with smoldering WM and were followed up for disease progression. Three patients remained asymptomatic on regular follow up and one received systemic chemotherapy on progression after four years.
Among twenty patients who received upfront treatment the indications for treatment were cytopenia in ten patients, constitutional symptoms in five, hyper viscosity in four and symptomatic lymphadenopathy in one patient. The treatment received were rituximab + cyclophosphamide + dexamethasone (RCD) in six, bendamustine + rituximab (BR) in three, bortezomib +rituximab + dexamethasone (BRD) in three, single agent rituximab in two, R-CHOP/ RCVP in two, cyclophosphamide + prednisolone (EP) in two, thalidomide + dexamethasone (TD) in one, bortezomib + dexamethasone (BD) in one patient. Sixteen patients received rituximab-based chemotherapy. Four patients with features with hyper viscosity underwent plasmapheresis.
Of the twenty patients who took treatment 13 patients (65%) attained CR, two (10%) attained PR, one patient (5%) progressed. One patient (5%) died during chemotherapy and three patients (15%) discontinued treatment. The patient who progressed on first line chemotherapy received BR as second line treatment and died while on chemotherapy. Three patients had relapsed with a median DFS of 12 months and received second line treatment with RCD/ BR/ lenalidomide + dexamethasone. The patient who received RCD as second line chemotherapy attained CR, relapsed again after 24 months for which she received BRD and continues to be in remission at 30 months. The other two patients defaulted treatment, One patient developed lung cancer at 88 months of follow up and another patient contracted COVID 19 infection and died due to COVID pneumonia.
Survival (Figure 2)
The PFS at 3 years was 69.4%. There was a significant difference in PFS with respect to presence of B symptoms, platelet count and monoclonal protein level in SPE.
Figure 2. a and b, Kaplan Meier Curve Showing PFS and OS at 3 Years.
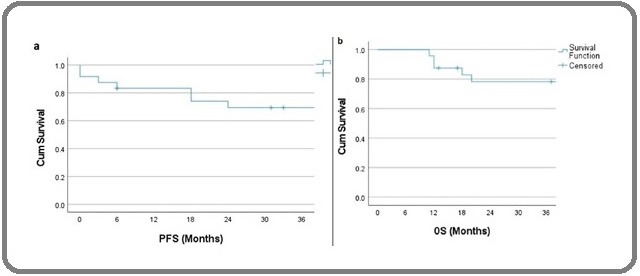
The 3-year OS was 78 %. OS for patients with age group < 65 years was 85.7 % and > 65 years was 75 with a P value of 0.47. There was no significant difference in OS with respect to age, sex, B symptoms, hemoglobin level, platelet count, serum LDH, monoclonal protein level in SPE, beta2 microglobulin (Table 3).
| Variables | PFS | OS | ||||||
| P-value | Hazard ratio (HR) | 95% CI for HR | P value | Hazard Ratio | 95% CI for HR | |||
| Lower | Upper | Lower | Upper | |||||
| Age | 0.677 | 0.737 | 0.175 | 3.098 | 0.488 | 2.142 | 0.249 | 18.391 |
| < 65 v/s > 65 years | ||||||||
| Sex | 0.677 | 1.357 | 0.323 | 5.701 | 0.488 | 0.467 | 0.054 | 4.009 |
| Male v/s Female | ||||||||
| B symptoms | 0.175 | 0.018 | 0 | 5.918 | 0.27 | 0.022 | 0 | 19.603 |
| Hemoglobin (g/dL) | 0.667 | 1.371 | 0.326 | 5.769 | 0.271 | 3.345 | 0.39 | 28.717 |
| >10 v/s < 10 | ||||||||
| Platelet Count (mm) 3 | 0.068 | 3.869 | 0.905 | 16.528 | 0.091 | 4.039 | 0.799 | 20.424 |
| 1 lakh v/s <1 lakh | ||||||||
| M Protein (g/dL) | 0.046 | 4.602 | 1.026 | 20.637 | 0.19 | 3.481 | 0.539 | 22.48 |
| <4 v/s > 4 | ||||||||
| IFE | 0.266 | 0.304 | 0.037 | 2.48 | 0.955 | 1.05 | 0.192 | 5.749 |
| IgM-k v/s IgM-λ | ||||||||
| Beta 2Microglobulin (g/dL) | 0.542 | 1.92 | 0.236 | 15.625 | 0.516 | 0.567 | 0.102 | 3.146 |
PFS, progression free survival; OS, Overall survival; CI, Confidence Interval; B symptoms, fever/weight loss/night sweats; M protein, Monoclonal band in serum protein electrophoresis; IFE, Immunofixation Electrophoresis; IgM-k, Immunoglobulin M- kappa; IgM- λ, Immunoglobulin M-Lambda.
The three-year OS was 75% in the group of patients who received rituximab+/- chemotherapy versus 50 % in the non-rituximab arm with a P value of 0.8.
Discussion
Waldenstrom macroglobulinemia or lymphoplasmacytic lymphoma is an extremely rare neoplasm. The disease was first described by Jan G. Waldenstrom in 1944, who reported an unusual presentation of lymphadenopathy, anaemia, bleeding, raised ESR, hyperviscosity and IgM hypergammaglobulinemia [9-11]. It is usually seen in the seventh or eighth decade of life with a male predominance. WM and LPL overlap, the only differentiating criteria being presence of IgM paraprotein in WM. In our study, WM constituted 0.68% of NHL with a median age at diagnosis of 67 years and male predominance.
The diagnosis of WML is usually challenging. About 25% of patients are asymptomatic at diagnosis, with 40% to 70% developing symptomatic disease within 3 years and 10 years of diagnosis [12]. The symptoms are caused by neoplastic infiltration of organs and elevated levels of IgM paraprotein. Patients usually present with constitutional symptoms like fatigue, fever, weight loss [13]. About one third of patients present with anaemia due to decreased erythropoiesis because of bone marrow infiltration, decreased erythrocyte survival due to IgM associated hemolysis, low erythropoietin levels, and iron deficiency [14]. Lymphadenopathy and or hepatosplenomegaly is seen in about 25% of cases and some may present with involvement extranodal sites such as skin, stomach, bowel etc [9,15-18]. WM can present with hyperviscosity symptoms in 30%, autoimmune hemolysis seen in 20%, coagulopathy and or diarrhoea. Symptoms related to hyperviscosity include headache, visual disturbances, neurologic symptoms and bleeding. Precipitation of IgM paraproteins can result in cryoglobulinemia causing Raynaud phenomena and cold urticaria [10, 19]. The autoimmue hemolysis is caused by cold agglutinins [12,16-18]. In the present study, 38% had constitutional symptoms, 50% had anaemia, 15% each had thrombocytopenia, lymphadenopathy, hepatosplenomegaly and symptoms of hyperviscosity. All our patients had a histological diagnosis of WM, 73% had IgM kappa and 27% had IgM lambda disease.
The 2017 WHO classification of tumors of hematopoietic and lymphoid tissues have established four diagnostic criteria for WM [11].
1. Presence of IgM monoclonal gammopathy
2. Infiltration of bone marrow by small lymphocytes showing plasmacytoid or plasma cell differentiation.
3. Bone marrow infiltration showing an intertrabecular pattern.
4. Immunophenotype supportive of WM/LPL including surface IgM+, CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, CD5 variable, CD10 -, CD23-, CD103-, CD108-
Most WM/LPL patients have monoclonal IgM, a minority can have both IgM and IgG.
The IPSS-WM is the most well developed and validated tool for risk stratification of symptomatic WM patients [3]. The staging system is based on five common clinical characteristics: age >65 years, haemoglobin ≤ 11.5gm/dL, platelets ≤ 100x109/L, β2 microglobulin>3mg/L, serum IgM > 7gm/dL. Low risk disease is defined as one or less adverse factor and age ≤ 65 years; intermediate risk disease defined as two adverse factors and age > 65 years; and high risk disease defined as having three or more adverse factors. Kastritis reported modified IPSS-WM system including LDH also as a risk factor to delineate high risk patients with a median survival of less than 3years [20]. Sex, B symptoms, IgM value, performance status, presence of hyperviscosity, bone marrow infiltration, and cytogenetic changes have also been reported as risk factors [21]. Rarely, WM can transform into an aggressive immunoblastic variant or a high grade lymphoma [11,19]. The most common genetic event in WM is an acquired mutation affecting the MYD88 gene is reported in 90% of patients. A second mutation affecting CXCR4 gene occurs in one third cases [22]. As per the IPSS- WP scoring, intermediate and high risk constituted 46.15% each and low risk constituted 7.7% in our patient group.
Management of WM is similar to other indolent small B cell lymphomas. Current therapeutic options in WM do not result in cure. The goal of treatment in symptomatic WM is to keep the disease under control as long as possible without impairing the quality of life. Treatment depends on patient symptoms and extent of end organ involvement. Patients with asymptomatic WM are managed by close observation. In symptomatic patients, the treatment of choice is to combine rituximab and chemotherapy [23]. Low risk symptomatic patients can be managed with single agent rituximab [24]. A combination of rituximab and bendamustine (BR) or cyclophosphamide and dexamethasone (DRC) is usually recommended [25,26]. The combination BR when compared to R CHOP showed statistically significant improvement in PFS (69.5 months versus 31.2 months) [26,27]. Rituximab monotherapy is considered in patients with poor general condition [28]. Ibrutinib has been approved as first line in patients not suitable for rituximab/ chemotherapy and in those with recurrences and those refractory to rituximab [29-31]. Bone marrow transplant is reserved for younger patients with extensive bone marrow involvement. If symptoms of hyperviscosity is present, plasmapheresis is recommended prior to chemotherapy [16, 32-34]. Eighty percent of the patients who were given systemic treatment received rituximab based chemotherapy in the present study.
The median survival of patients with WM is about 7.4 years, mortality is associated with symptomatic disease, whereas asymptomatic patients have survival similar to the general population [33]. The 5 year survival rates for IPSS-WM low risk, intermediate risk and high risk are 87%,68% and 36% respectively [3]. The absence of MYD 88 mutation is associated with poor outcome. WM that transforms to diffuse large B cell lymphoma is also associated with poor survival [5,17,19,33, 35]. In the present study, the 3 year PFS was 69.4% and OS was 78%.
In conclusion, WM is a low grade B cell lymphoproliferative disorder having an indolent course and do not require treatment for prolonged period. Diagnosis is challenging due to lack of distinct diagnostic features. Rituximab containing regimens are the standard of care. Newer targeted treatment options may improve the outcome of this incurable disease.
Acknowledments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for- profit sectors. The authors declare no conflict of interest
References
- Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: a large population-based study Wang H, Chen Y, Li F, Delasalle K, Wang J, Alexanian R, Kwak L, et al . Cancer.2012;118(15). CrossRef
- Diagnosis and management of Waldenstrom's macroglobulinemia Dimopoulos MA , Kyle RA , Anagnostopoulos A, Treon SP . Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology.2005;23(7). CrossRef
- International prognostic scoring system for Waldenstrom macroglobulinemia Morel P, Duhamel A, Gobbi P, Dimopoulos MA , Dhodapkar MV , McCoy J, Crowley J, et al . Blood.2009;113(18). CrossRef
- Monoclonal IgM Gammopathy and Waldenström's Macroglobulinemia Grunenberg A, Buske C. Deutsches Arzteblatt International.2017;114(44). CrossRef
- Waldenström macroglobulinemia: What a hematologist needs to know Kapoor P, Paludo J, Vallumsetla N, Greipp PR . Blood Reviews.2015;29(5). CrossRef
- Amyloidosis and Waldenström's macroglobulinemia Gertz MA , Merlini G, Treon SP . Hematology. American Society of Hematology. Education Program.2004. CrossRef
- Novel aspects pertaining to the relationship of Waldenström's macroglobulinemia, IgM monoclonal gammopathy of undetermined significance, polyclonal gammopathy, and hypoglobulinemia McMaster ML , Kristinsson SY , Turesson I, Björkholm M, Landgren O. Clinical Lymphoma & Myeloma.2009;9(1). CrossRef
- Report from the Sixth International Workshop on Waldenström's Macroglobulinemia Treon SP , Merlini G, Morra E, Patterson CJ , Stone MJ . Clinical Lymphoma, Myeloma & Leukemia.2011;11(1). CrossRef
- Waldenström macroglobulinemia: 2015 update on diagnosis, risk stratification, and management Gertz MA . American Journal of Hematology.2015;90(4). CrossRef
- Lymphoplasmacytic lymphoma and Waldenström macroglobulinemia Naderi N, Yang DT . Archives of Pathology & Laboratory Medicine.2013;137(4). CrossRef
- The 2016 revision of the World Health Organization classification of lymphoid neoplasms Swerdlow SH , Campo E, Pileri SA , Harris NL , Stein H, Siebert R, Advani R, et al . Blood.2016;127(20). CrossRef
- Updates in prognostication and treatment of Waldenström's macroglobulinemia Advani P, Paulus A, Ailawadhi S. Hematology/Oncology and Stem Cell Therapy.2019;12(4). CrossRef
- Waldenström's macroglobulinemia: clinical features, complications, and management Dimopoulos MA , Panayiotidis P, Moulopoulos LA , Sfikakis P, Dalakas M. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology.2000;18(1). CrossRef
- Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases García-Sanz R, Montoto S, Torrequebrada A, Coca AG , Petit J, Sureda A, Rodríguez-García JA , et al . British Journal of Haematology.2001;115(3). CrossRef
- Waldenström Macroglobulinemia: Review of Pathogenesis and Management Yun S, Johnson AC , Okolo ON , Arnold SJ , McBride A, Zhang L, Baz RC , Anwer F. Clinical Lymphoma, Myeloma & Leukemia.2017;17(5). CrossRef
- Rituximab intolerance in patients with Waldenström macroglobulinaemia Castillo JJ , Kanan S, Meid K, Manning R, Hunter ZR , Treon SP . British Journal of Haematology.2016;174(4). CrossRef
- Clinicopathologic features and outcomes of lymphoplasmacytic lymphoma patients with monoclonal IgG or IgA paraprotein expression Cao X, Medeiros LJ , Xia Y, Wang X, Thomas SK , Loghavi S, Li X, et al . Leukemia & Lymphoma.2016;57(5). CrossRef
- Initial Evaluation of the Patient with Waldenström Macroglobulinemia Castillo JJ , Treon SP . Hematology/Oncology Clinics of North America.2018;32(5). CrossRef
- Biology, prognosis, and therapy of Waldenström Macroglobulinemia Castillo JJ , Ghobrial IM , Treon SP . Cancer Treatment and Research.2015;165. CrossRef
- Competing risk survival analysis in patients with symptomatic Waldenström macroglobulinemia: the impact of disease unrelated mortality and of rituximab-based primary therapy Kastritis E, Kyrtsonis M, Morel P, Gavriatopoulou M, Hatjiharissi E, Symeonidis AS , Vassou A, et al . Haematologica.2015;100(11). CrossRef
- Waldenström macroglobulinemia. Development of diagnostic criteria and identification of prognostic factors Owen RG , Barrans SL , Richards SJ , O'Connor SJ , Child JA , Parapia LA , Morgan GJ , Jack AS . American Journal of Clinical Pathology.2001;116(3). CrossRef
- Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia Treon SP , Cao Y, Xu L, Yang G, Liu X, Hunter ZR . Blood.2014;123(18). CrossRef
- Waldenstrom's macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up Buske C, Leblond V, Dimopoulos M, Kimby E, Jäger U, Dreyling M. Annals of Oncology: Official Journal of the European Society for Medical Oncology.2013;24 Suppl 6. CrossRef
- Diagnosis and Management of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) Guidelines 2016 Kapoor P, Ansell SM , Fonseca R, Chanan-Khan A, Kyle RA , Kumar SK , Mikhael JR , et al . JAMA oncology.2017;3(9). CrossRef
- Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenström macroglobulinemia Paludo J, Abeykoon JP , Shreders A, Ansell SM , Kumar S, Ailawadhi S, King RL , et al . Annals of Hematology.2018;97(8). CrossRef
- Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: final analysis of a phase 2 study Kastritis E, Gavriatopoulou M, Kyrtsonis M, Roussou M, Hadjiharissi E, Symeonidis A, Repoussis P, et al . Blood.2015;126(11). CrossRef
- Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide Dimopoulos MA , Anagnostopoulos A, Kyrtsonis M, Zervas K, Tsatalas C, Kokkinis G, et al . Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology.2007;25(22). CrossRef
- Extended rituximab therapy in Waldenström's macroglobulinemia Treon SP , Emmanouilides C, Kimby E, Kelliher A, Preffer F, Branagan AR , Anderson KC , Frankel SR . Annals of Oncology: Official Journal of the European Society for Medical Oncology.2005;16(1). CrossRef
- Ibrutinib in previously treated Waldenström's macroglobulinemia Treon SP , Tripsas CK , Meid K, Warren D, Varma G, Green R, et al . The New England Journal of Medicine.2015;372(15). CrossRef
- Ibrutinib for patients with rituximab-refractory Waldenström's macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial Dimopoulos MA , Trotman J, Tedeschi A, Matous JV , Macdonald D, Tam C, Tournilhac Ol, et al . The Lancet. Oncology.2017;18(2). CrossRef
- New developments in the management of Waldenström macroglobulinemia Abeykoon JP , Yanamandra U, Kapoor P. Cancer Management and Research.2017;9. CrossRef
- Waldenström macroglobulinemia treatment algorithm 2018 Gertz MA . Blood Cancer Journal.2018;8(4). CrossRef
- Waldenstrom macroglobulinemia: prognosis and management Oza A, Rajkumar SV . Blood Cancer Journal.2015;5(3). CrossRef
- BCR pathway inhibition as therapy for chronic lymphocytic leukemia and lymphoplasmacytic lymphoma Wiestner A. Hematology. American Society of Hematology. Education Program.2014;2014(1). CrossRef
- Incidence of secondary malignancies among patients with Waldenström macroglobulinemia: An analysis of the SEER database Castillo JJ , Olszewski AJ , Hunter ZR , Kanan S, Meid K, Treon SP . Cancer.2015;121(13). CrossRef
License

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
Copyright
© Asian Pacific Journal of Cancer Care , 2024
Author Details
How to Cite
- Abstract viewed - 0 times
- PDF (FULL TEXT) downloaded - 0 times
- XML downloaded - 0 times